
Note
Go to the end to download the full example code.
Equilibrating a box of acetonitrile with MD#
Goals#
In this tutorial we learn how to perform a thermal equilibration of a box of acetonitrile molecules using ASE. We will:
build a coarse-grained CH₃–C≡N triatomic model.
set up a periodic box at the experimental density (298 K).
apply rigid-molecule constraints.
use the ACN force field.
equilibrate with Langevin dynamics.
scale small box of 27 molecules to 216 molecules.
ACN model#
The acetonitrile force field implemented in ASE (ase.calculators.acn)
is an interaction potential between three-site linear molecules. The atoms
of the methyl group are treated as a single site centered on the methyl
carbon (hydrogens are not explicit). Therefore:
assign the methyl mass to the outer carbon (
m_me),use the atomic sequence Me–C–N repeated for all molecules,
keep molecules rigid during MD with
FixLinearTriatomic.
import matplotlib.pyplot as plt
import numpy as np
import ase.units as units
from ase import Atoms
from ase.calculators.acn import ACN, m_me, r_cn, r_mec
from ase.constraints import FixLinearTriatomic
from ase.io import Trajectory
from ase.md import Langevin
from ase.visualize.plot import plot_atoms
Step 1: Molecule#
Build one CH3–C≡N as a linear triatomic “C–C–N”. The first carbon is the methyl site; we assign it the CH3 mass. Rotate slightly to avoid perfect alignment with the cell axes.
pos = [[0, 0, -r_mec], [0, 0, 0], [0, 0, r_cn]]
atoms = Atoms('CCN', positions=pos)
atoms.rotate(30, 'x')
masses = atoms.get_masses()
masses[0] = m_me
atoms.set_masses(masses)
mol = atoms.copy()
mol.set_pbc(False)
mol.set_cell([20, 20, 20])
mol.center()
fig, ax = plt.subplots(figsize=(4, 4))
plot_atoms(
mol,
ax=ax,
rotation='30x,30y,0z',
show_unit_cell=0,
radii=0.75,
)
ax.set_axis_off()
plt.tight_layout()
plt.show()
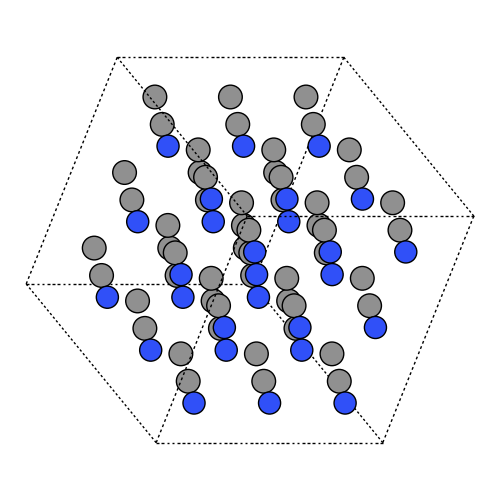
Step 2: Set up small box of 27-molecules#
Match density 0.776 g/cm^3 at 298 K. Compute cubic box length L from mass and density. Build 3×3×3 supercell and enable PBC.
density = 0.776 / 1e24 # g / Å^3
L = ((masses.sum() / units.mol) / density) ** (1 / 3.0)
atoms.set_cell((L, L, L))
atoms.center()
atoms = atoms.repeat((3, 3, 3))
atoms.set_pbc(True)
box27 = atoms.copy()
fig, ax = plt.subplots(figsize=(5, 5))
plot_atoms(
box27,
ax=ax,
rotation='35x,35y,0z',
show_unit_cell=2,
radii=0.75,
)
ax.set_axis_off()
plt.tight_layout()
plt.show()
Step 3: Set constraints#
Keep each “C–C–N” rigid during MD using FixLinearTriatomic.
Step 4: MD run for 27-molecules system#
Assign ACN with cutoff = half the smallest box edge. Langevin MD at 300 K, 2 fs timestep. Save a frame every step.
Note
This example uses a relatively large timestep to demonstrate the usage of the code. In general, a smaller timestep should be used for this molecular dynamics simulation. Generally, the timestep depends on the chemical system, its constraints and is in the order of 0.5-2~fs.
atoms.calc = ACN(rc=np.min(np.diag(atoms.cell)) / 2)
tag = 'acn_27mol_300K'
md = Langevin(
atoms,
2 * units.fs,
temperature_K=300,
friction=0.01,
logfile=tag + '.log',
)
traj = Trajectory(tag + '.traj', 'w', atoms)
md.attach(traj.write, interval=50) # writing the structure every 100 fs
md.run(10) # 10 timestseps @ 2 fs = 0.02 ps
/home/ase/.local/lib/python3.13/site-packages/ase/md/langevin.py:110: FutureWarning: The implementation of `fixcm=True` in `Langevin` does not strictly sample the correct NVT distributions. The deviations are typically small for large systems but can be more pronounced for small systems. Use `fixcm=False` together with `ase.constraints.FixCom`. `fixcm` is deprecated since ASE 3.28.0 and will be removed in a future release.
warnings.warn(msg, FutureWarning)
True
Step 4: Monitoring a MD simulation#
Tracking different properties such as the temperature of a system makes sense to monitor the behavior of a system and insure correct physics.
times_ps, epots, ekins, etots, temps = [], [], [], [], []
sample_interval = 10 # sample every 10 MD steps for lighter plots
def sample():
# Time in ps (same as MDLogger: dyn.get_time() / (1000 * units.fs))
t_ps = md.get_time() / (1000.0 * units.fs)
ep = atoms.get_potential_energy() # eV total
ek = atoms.get_kinetic_energy() # eV total
T = atoms.get_temperature() # K
times_ps.append(t_ps)
epots.append(ep)
ekins.append(ek)
etots.append(ep + ek)
temps.append(T)
# initial sample at t=0
sample()
md.attach(sample, interval=sample_interval)
md.run(1500) # 1500 timesteps @ 2 fs = 3 ps
True
Plot Instantaneous temperature vs time. Does the system equilibrated well? What is the average temperature? Should we run longer simulations?
fig, ax = plt.subplots(figsize=(6, 4))
# line at 300 K for comparison
ax.axhline(300, color='black', linestyle='--')
ax.plot(times_ps, temps, label='T (K)')
ax.set_xlabel('Time (ps)')
ax.set_ylabel('Temperature (K)')
ax.legend(loc='best')
ax.grid(True, linewidth=0.5, alpha=0.5)
plt.tight_layout()
plt.show()
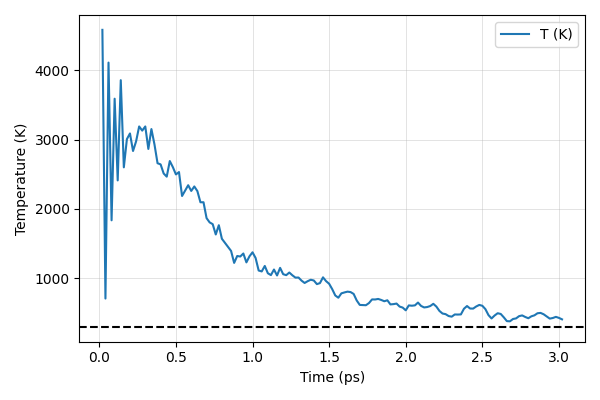
Hint: How does the temperature of the atoms align with the black dashed line at 300 K? Try running for 5 ps and 10 ps, is the system equilibrated then?
Step 6: Scale system size to 216 molecules#
Repeat 2×2×2 to reach 216 molecules. Reapply constraints and update the ACN cutoff for the new cell.
Step 7: MD run for 216-molecules system#
tag = 'acn_216mol_300K'
md = Langevin(
atoms,
2 * units.fs,
temperature_K=300,
friction=0.01,
logfile=tag + '.log',
)
traj = Trajectory(tag + '.traj', 'w', atoms)
md.attach(traj.write, interval=50)
times_ps, epots, ekins, etots, temps = [], [], [], [], []
sample_interval = 10 # sample every 10 MD steps for lighter plots
# initial sample at t=0
sample()
md.attach(sample, interval=sample_interval)
md.run(10) # 10 timesteps @ 2 fs = 0.02 ps
True
This simulation is only run very briefly to show how to scale a simulation. Try running the simulation longer and plot the temperature and other properties. Does this size take longer to equilibrate?
Next steps#
View trajectories:
Plot other thermodynamic quantities (p.e., k.e., and total energy).